Overview
The primary goal of the Evolutionary Dynamics Lab is to contribute to our understanding of how malignant traits evolve in cancer. The verb “evolve” is absolutely correct here. Cancer arises as originally normal cells acquire “hallmark” characteristics of malignancy (to use Douglas Hanahan and Robert Weinberg’s [2000, 2011] justifiably famous term). Overwhelming evidence shows that these characteristics (phenotypes) arise via alterations in the hereditary material (genetic, epigenetic and gross genomic derangements). Therefore, hallmark phenotypes are inherited. As tumors expand, cell lineages (clones) accumulate heritable differences because further genetic changes occur during replication. Not all cells in the tumor accumulate the same genetic alterations because these arise in single cells residing in a tumor of many cells. Only the descendants of the original mutant (the clone) inherit the mutation. As a result, tumors comprise a variety of genetically distinct clones all competing for resources. Certain clones, due to their heritable characteristics, will compete better than others and pass that advantage on to their descendants. Advantaged clones will expand faster than others, altering the gross tumor’s biological and clinical behavior. This process is precisely evolution by natural selection as envisioned by Darwin. Our goal is to develop a correct mathematical theory of this process. Such a theory would enlighten our understanding of the biology of this disease and, hopefully, contribute to personalized treatments.
Projects
Project 1: Hypertumors: Cancer Gets Cancer
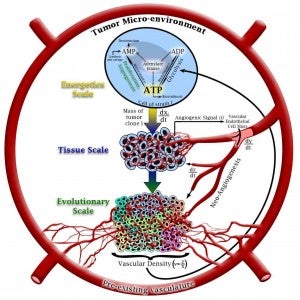 In 2004 we proposed that free-rider clones within a tumor can grow as a tumor-on-a-tumor, which we termed “hypertumor,” analogous to the established term, hyperparasite–a parasite of a parasite (Nagy 2004). Our current model includes intracellular (energetic), tissue (tumor) and evolutionary scales; see figure (right, by Scott Bickel. See Bickel et al. 2014).
In 2004 we proposed that free-rider clones within a tumor can grow as a tumor-on-a-tumor, which we termed “hypertumor,” analogous to the established term, hyperparasite–a parasite of a parasite (Nagy 2004). Our current model includes intracellular (energetic), tissue (tumor) and evolutionary scales; see figure (right, by Scott Bickel. See Bickel et al. 2014).
Collaborators: Dieter Armbruster, Fabio Milner, Robert Alvarez
Current Students: Scott Bickel
Former Students: Joe Juliano, Erin Victor, Jenese Cropper, Kelly Thompson
Project 2: Androgen Resistance in Prostate Cancer
It has long been known that cells in the most common form of prostate cancer depend on androgen (testosterone, DHT). Therefore, standard treatment for advanced prostate tumors calls for androgen ablation–essentially castration, either chemical or physical–to reduce serum testosterone concentration. At treatment onset, tumors invariably shrink. However, they nearly equally invariably evolve away from androgen dependence to become castration resistant. We have deployed a number of models of this evolutionary process in an attempt to understand why it occurs and find novel ways to avoid, or at least delay, it. We base most of the models in this project on the Droop formalism, which has become a very important model in ecology (see project 3 description).
Collaborator: Yang Kuang
Current Students: Jason Morken
Former Students: Travis Portz, Aaron Packer (PhD 2014), Rebecca Everett (PhD 2015)
Project 3: Evolutionary ecology of cancer: resource ratio theory
Biological stoichiometry, or resource ratio theory, has become established as one of the more general theories in biology. It exploits the fact that living things are built of elements in nearly fixed ratios, an insight that greatly simplifies growth equations from cells to populations to communities and more. While studying ratios of P:N:C in a sampling of cancer types, we discovered an interesting pattern–some tumors are rich in P and some are not (Elser et al. 2007). P-rich tumors arise in tissues more exposed to insult from the outside world (lung, colon), while P-poor tumors are found in “protected” tissues (kidney, liver). We hypothesize that P-rich tumors act like invasive “r-selected” ecological species, growing primarily by increasing proliferation rates, since demand for P increases in rapidly dividing cells (to support upregulation of ribosomes to support biosynthesis). P-poor tumors, then, would be more “K-selected,” becoming better competitors and growing primarily by resisting apoptosis and necrosis. Our modeling supports these notions, but we have yet to find funding to perform experiments to test it.
Collaborators: Jim Elser, Yang Kuang
Current Students: David Ung, Kirsten Karr
Former Students: Daniel Stegman, Karl Lundin